BCIC分子预测大模型平台
告别盲目试错,帮您从千万种可能中快速找到结果
核心工作流
通过四大核心步骤,实现从预测到验证的完整闭环,高效推动分子研究进程。
性质预测
预测物理化学及生物活性
结构生成
设计具特定性质的新分子
反应预测
推断反应路径、产物及条件
构效关系
建立结构与性质的定量关系
三大核心模块
数据智能处理
整合蛋白质序列、PDB库等数十亿级数据源,构建全面知识图谱。
高速分子模拟
基于独有算法驱动分子动力学模拟演算,高效探索构象空间。
三维可视化呈现
动态展示分子结合模式与构象变化,提供直观的交互分析体验。
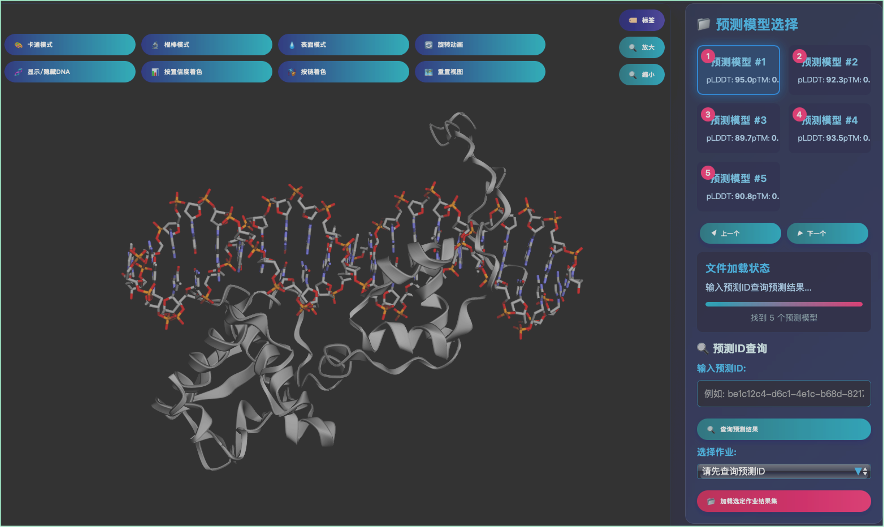
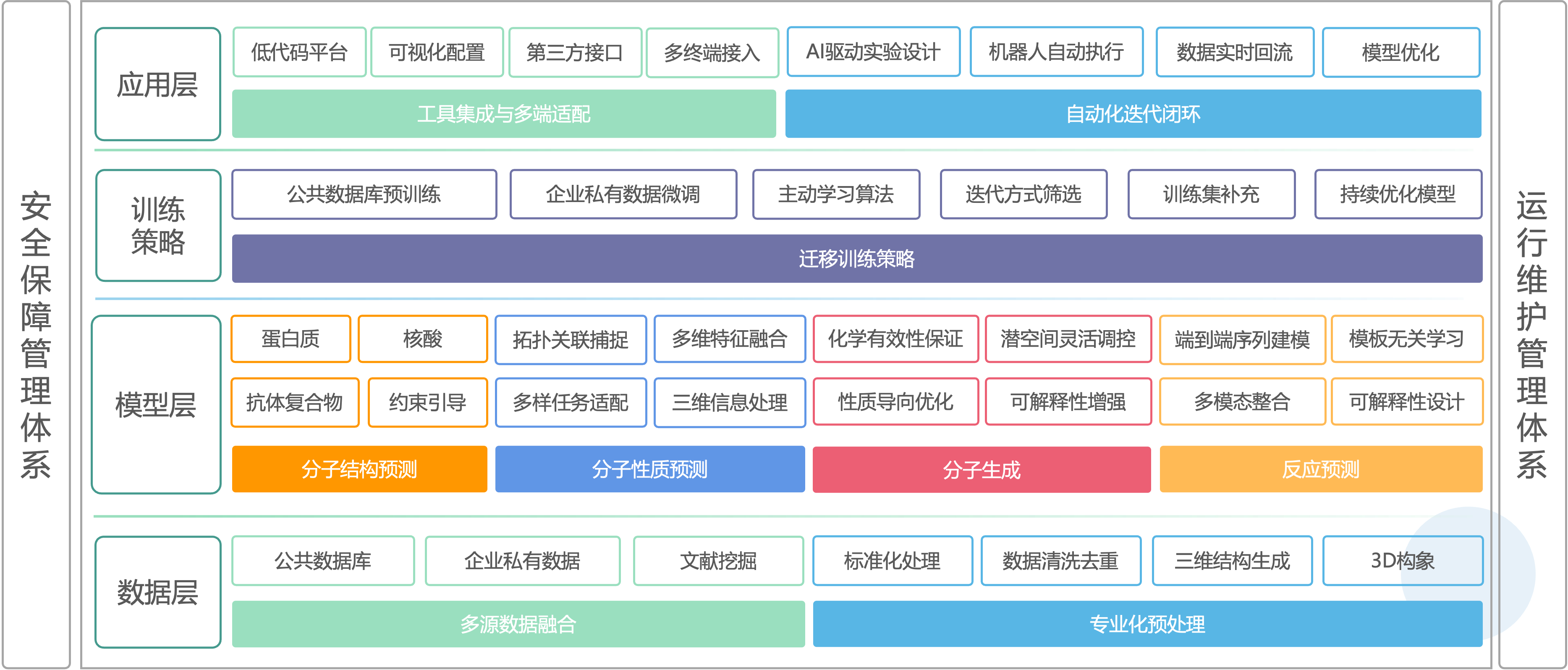
平台技术架构
核心技术优势
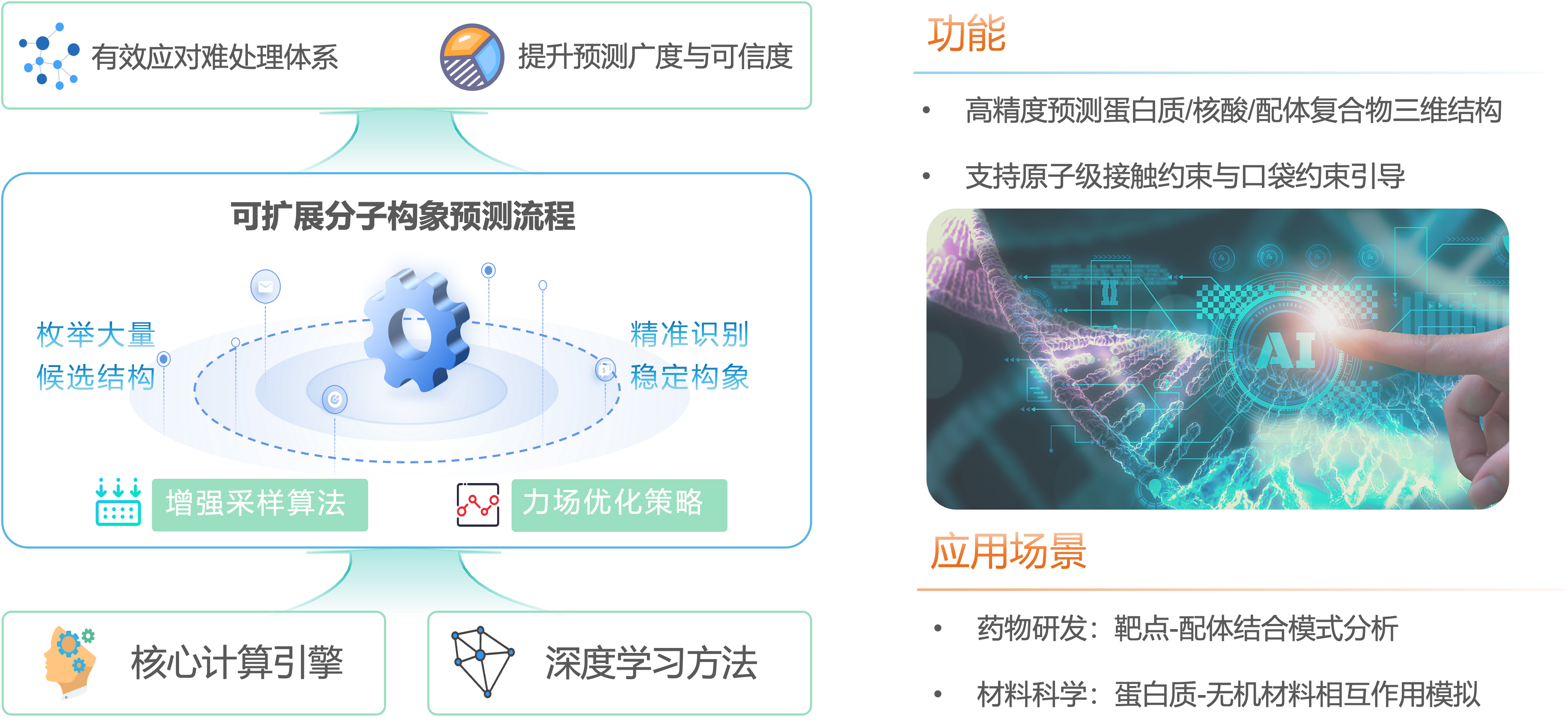
智能算法驱动的高效分子结构预测体系
依托核心计算引擎及深度学习方法,我们构建了一套可扩展的分子构象预测流程,有效应对柔性分子、大环化合物等难处理体系。通过引入增强采样算法与力场优化策略,系统能够在极短时间内枚举大量候选构并精准识别稳定构象,极大提升了药物设计与材料开发中结构预测的覆盖广度与可信度。
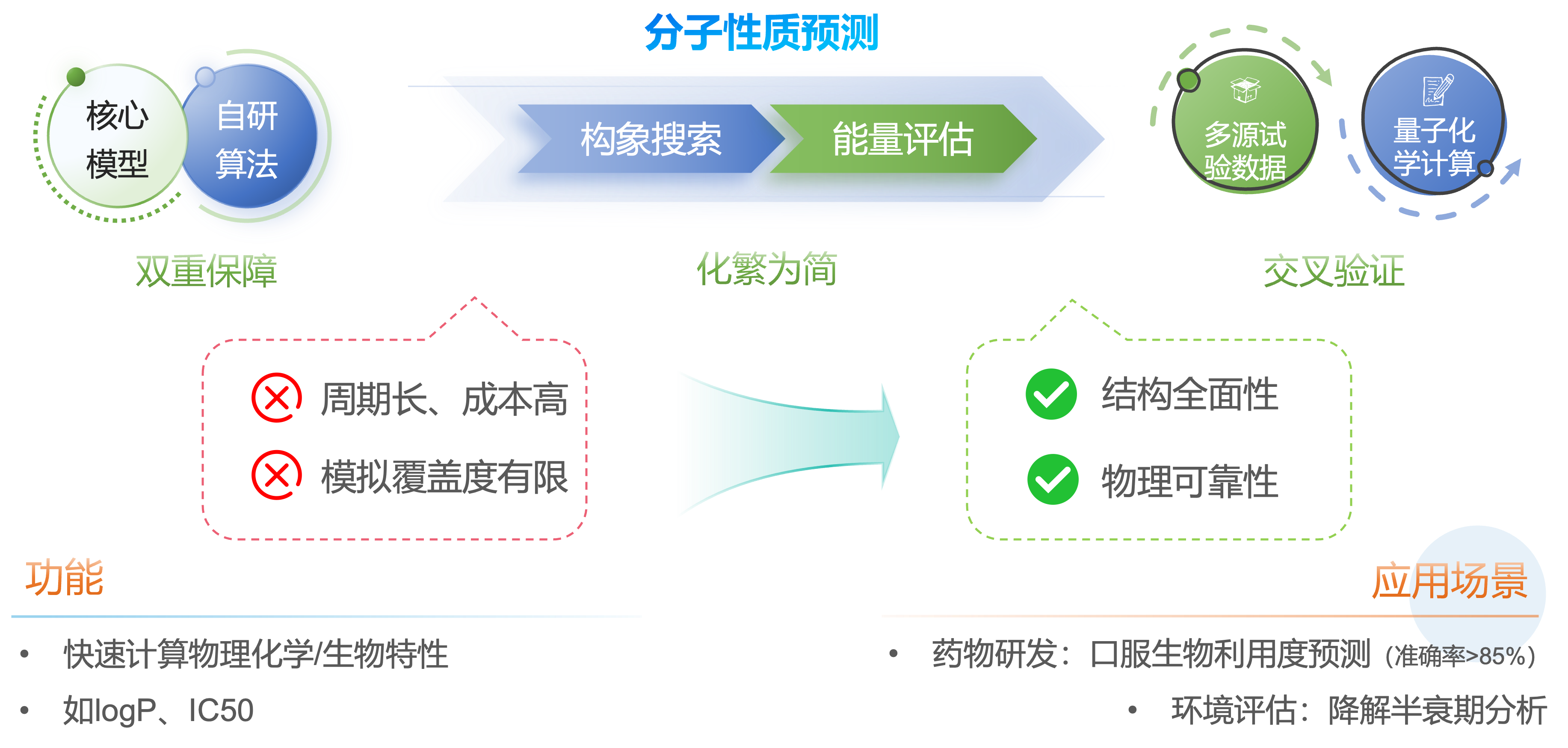
双重技术保障的精准分子性质预测系统
通过集成成分子预测核心模型与自研算法,我们实现了对复杂分子体系的高效构象搜索与能量评估,显著克服了传统实验方法周期长、成本高以及计算模拟覆盖度有限的问题。系统能够快速筛选出能量最低的“优势构象”,并结合多源实验数据与量子化学计算进行交叉验证,确保预测结果兼具结构全面性与物理可靠性。
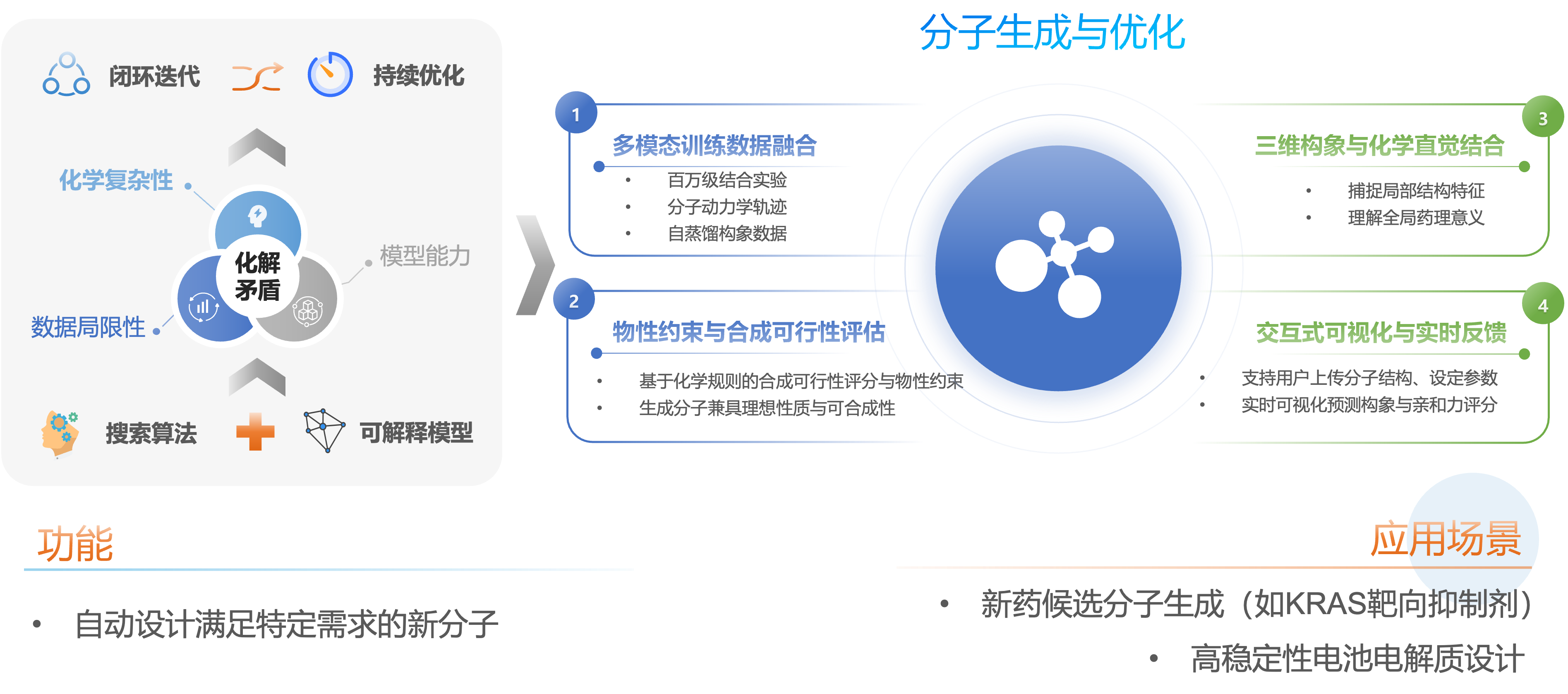
透明可视的可解释分子生成与优化平台
通过构建更精准的性质预测模型和可合成性约束,开发更高效的搜索算法、更透明的可解释模型,化解“化学复杂性”、“数据局限性”与“模型能力”之间的矛盾,加速实验与模型的闭环迭代。优化包括:多模态的约束与合成可行性评估、三维构象与化学直觉结合、交互式可视化与实时反馈。
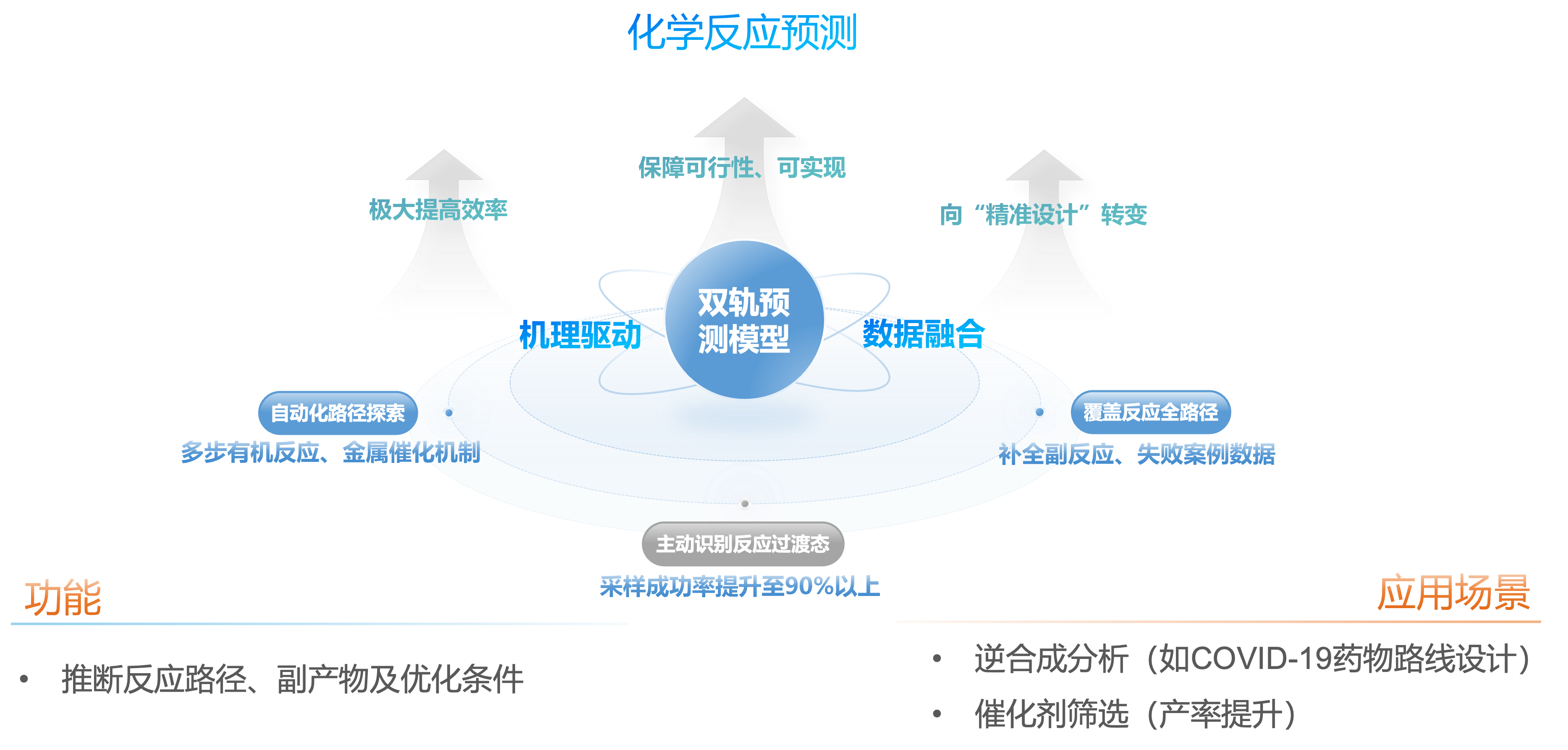
全路径探索的自动化化学反应预测机制
结合量子化学计算与机器学习算法,我们构建了“机理驱动-数据融合”的双轨预测模型。该模型通过引入高精度量子力学方法(如GFN2-xTB/DFT)与大语言模型辅助的化学逻辑推理,实现了对多步有机反应、金属催化机制(如DFT催化)的自动化路径探索,系统能够主动识别反应过渡态(TS),采样成功率提升至90%以上,并补全副反应、失败案例数据,构建覆盖反应全路径的势能面(PES)图谱。
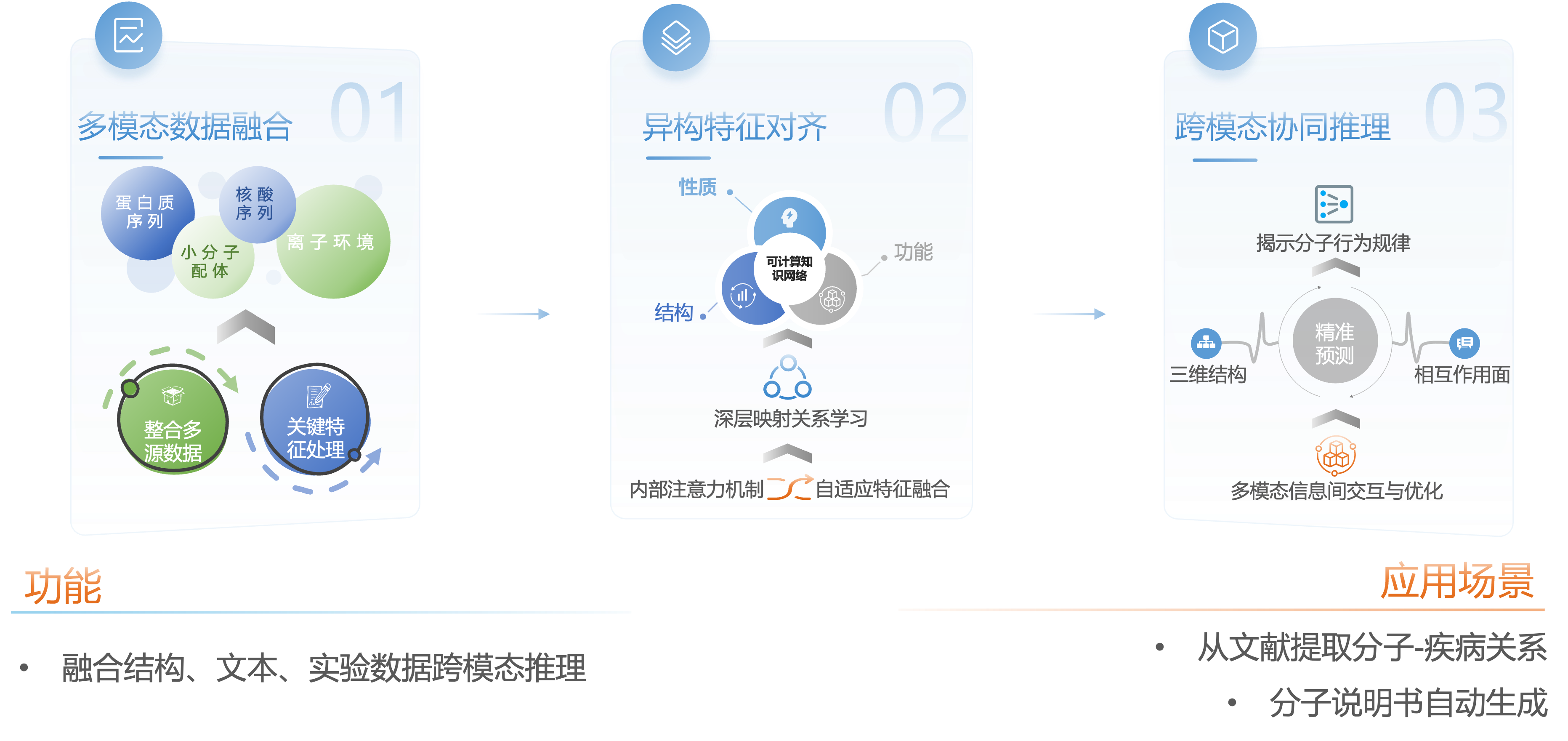
多模态融合驱动的分子理解策略
依托分子预测核心模型,构建了“数据融合—特征对齐—跨模态推理”的完整技术链条:多模態数据融合;协同处理包括蛋白质序列、核酸序列、小分子配体的化学结构乃至离子环境等多种输入信息。特征异构特征对齐:模型在异构特征空间中对齐不同模态的分子信息。跨模态协同推理:利用扩散网络等先进算法,模型进行精细的跨模态推理生成。